In a typical synthesis of Cu3 N nanocubes, 0.06 g Cu (NO3 )2·3H2 O was dissolved in the mixture of 5 mL 1-octadecylamine and 5 mL 1-octadecene. The solution was degassed for at least 1 hr and then heated at 150 °C for more than 3 h under magnetic stirring. During the reaction, the color of the solution changed from blue to green, and finally to yellow. The solution temperature was then raised to 250 °C at a rate of approximately 5 °C/min and kept for 30 min. The product was collected by centrifugation, washed with alcohol several times and finally dispersed in chloroform.
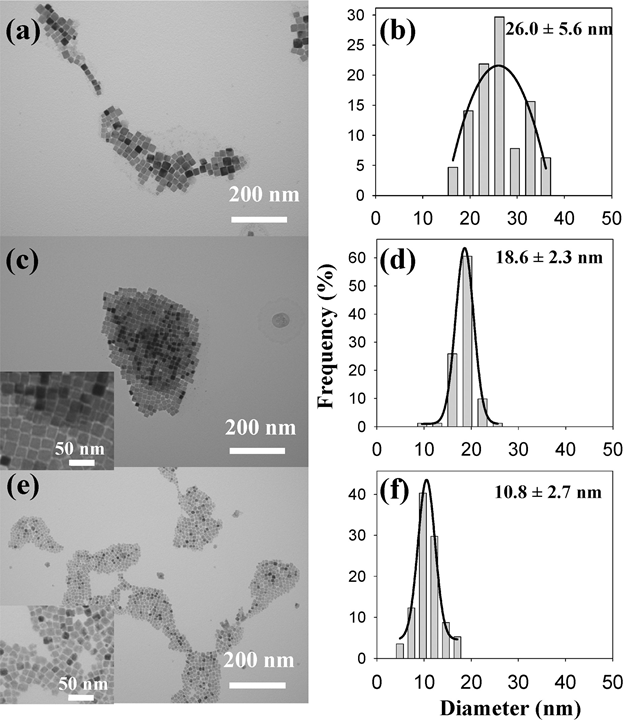
Figure. TEM images and the corresponding crystal size histograms of Cu3N nanocrystals synthesized with different organic solvents: (a,b) octadecylamine and octadecene, (c,d) hexadecylamine and octadecene,and (e,f) oleylamine and octadecene[J. Am. Chem. Soc. 2011, 133, 15236–15239]
Figure. (top) TEM image of the as-prepared Cu3N nanoparticles with pure oleylamine as capping agent. (bot) TEM images of Cu3N nanoparticles synthesized in oleylamine and octadecene. The nanoparticles were obtained by cooling the reaction solution suddenly from high temperature down to room temperature[J. Am. Chem. Soc. 2011, 133, 15236–15239]
1.2. Nanocrystal Cu3N
A high-temperature, high-pressure stainless steel reactor (125 mL, Parr Instruments Model 4752, 3000 psi maximum pressure) was loaded in an argon-filled glovebox with anhydrous copper chloride powder and a stoichiometric amount of sodium azide. The solid reagents were separately ground with a mortar and pestle and used as fine powders. The reactor containing the powder mixture and a Teflon-coated stir bar was partially filled with degassed toluene or THF (∼85 mL or 77% reactor fill) using inert atmosphere Schlenk transfer techniques. The reactor was sealed under an atmospheric pressure flow of nitrogen gas, placed in beaker-shaped custom heating mantle, and slowly heated with constant stirring using an external magnetic stirrer. Typically, the Cu3N precursors were heated from room temperature to an intermediate temperature (140 °C for toluene and 120 °C for THF). Heating rates included ramping to ∼50 °C (1−2 °C/min) and holding at this level for 4 h and then ramping to ∼100 °C and holding at this temperature overnight (10−12 h) to facilitate the synthesis of metal azide intermediates. After metal azide formation, subsequent temperature increases in ∼5−10 °C increments were made over several days (∼40 °C/day for toluene and ∼25 °C/day for THF) in order to facilitate moderate (nonexplosive) copper azide decomposition, as indicated by gas evolution rates monitored using the reactor’s analogue pressure gauge. Increases in reaction temperature were only made after the vessel pressure had reached a plateau, which usually occurred within a few hours. The maximum reaction temperature was determined as the point where gas evolution ceased, which indicated that all of the copper azide had decomposed. The maximum Cu3N reaction temperature (185 °C for toluene and THF) was typically maintained for nearly 1 day. The total solvothermal reaction time was 3−5 days and no significant product advantages were observed from longer reaction times.
Inorg. Chem. 2005, 44, 21, 7385–7393
Figure. SEM images of Cu3N products from copper azide decomposition in toluene (A, B) and in THF (C, D).
1.3. Ultrasmall Cu3N
In a glovebox under argon atmosphere (O2 < 0.1 ppm and H2O < 0.1 ppm), 50 mg of Cu(OMe)2 was added into a 10 mL glass vial. Afterward 5 mL benzylamine was added and the vessel was sealed with a Teflon cap and taken out of the glovebox. The reaction vessel was transferred into a preheated oil bath set at 140 °C and held at that temperature for 15 min under vigorous magnetic stirring. During heating, after about 3 min, the color of the reaction solution changed from dark blue to red. Afterward, the reaction mixture was cooled down to room temperature. The product was precipitated by adding 40 mL of hexane and collected by centrifugation at 4000 rpm for 15 min. The product was washed two more times using 40 mL of hexane and finally dried under nitrogen flux. The yield of the reaction was >90% with respect to Cu(OMe)2. To obtain stable colloidal dispersions of Cu3N nanoparticles, the wet products obtained after washing were directly dispersed in chloroform, THF, and NMP using ultrasonication for 10 min. The nanoparticle dispersion in chloroform was used for TEM sample preparation.
Chem. Mater. 2015, 27, 24, 8282–8288
2. Material Properties
2.1. XRD
asAS
ASAS
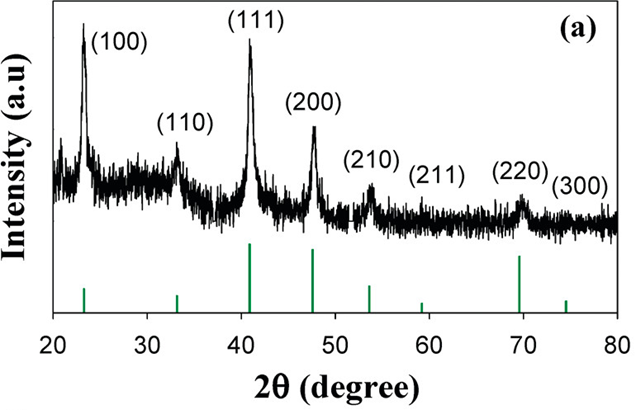
Fig. XRD pattern of the Cu3N nanocubes[J. Am. Chem. Soc. 2011, 133, 15236–15239]
Fig. XRD pattern of the solvothermal decomposition products from the reaction of CuCl2 and NaN3 in toluene at 185 °C (a) as-synthesized [NaCl peaks *] and (b) after washing with methanol [cubic Cu3N peaks] and (c) a methanol-washed product from the reaction of CuCl2 and NaN3 in THF at 185 °C[Inorg. Chem. 2005, 44, 21, 7385–7393]
a
2.2. XPS
asas
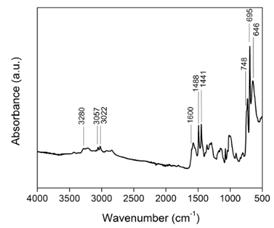
2.3. FT-IR
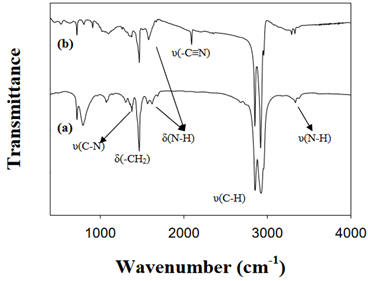
Figure. FT-IR spectra of (a) 1-octadecylamine and (b) as-prepared Cu3N nanocubes [J. Am. Chem. Soc. 2011, 133, 15236–15239]
Figure. FT-IR spectra of methanol-washed products from Cu3N synthesized in (a) toluene and (b) THF. The * indicates likely positions for solvent-based vibrations. [Inorg. Chem. 2005, 44, 21, 7385–7393]
Figure. [Chem. Mater. 2015, 27, 24, 8282–8288]
assa
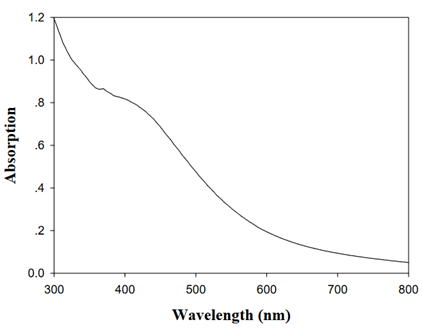
2.4. UV-Vis spectra
as
Figure. UV-Vis spectra of the Cu3N nanocubes in chloroform[J. Am. Chem. Soc. 2011, 133, 15236–15239]
as
3. Electrochemical Performance
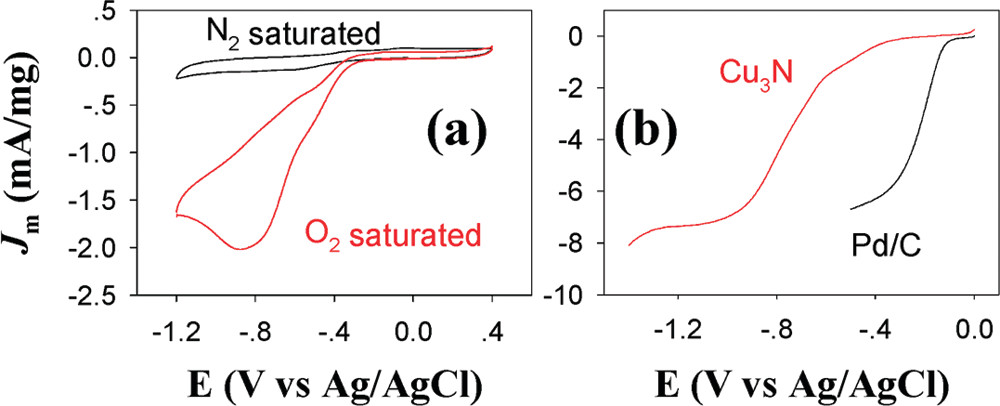
3.1. in Aqueous Solution
as
Figure. (a) CV of the Cu3N nanocubes in 0.1 M KOH solution. Scan rate 0.1V/s. (b) Rotating disk voltammograms in 0.1 M KOH solution saturated with oxygen (3025 rpm; scan rate, 5 mV s–1).[J. Am. Chem. Soc. 2011, 133, 15236–15239]
as
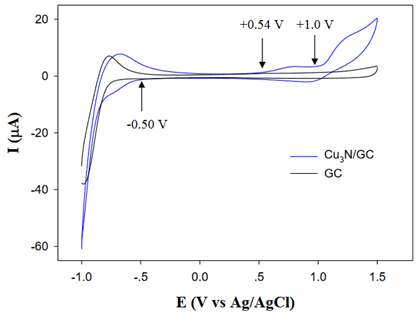
3.2. in NonAqueous Solution
as
Figure. CV of the Cu3N nanocubes in acetonitrile with 0.1 M tetrabutylammonium perchlorate. Scan rate 0.1V/s[J. Am. Chem. Soc. 2011, 133, 15236–15239]


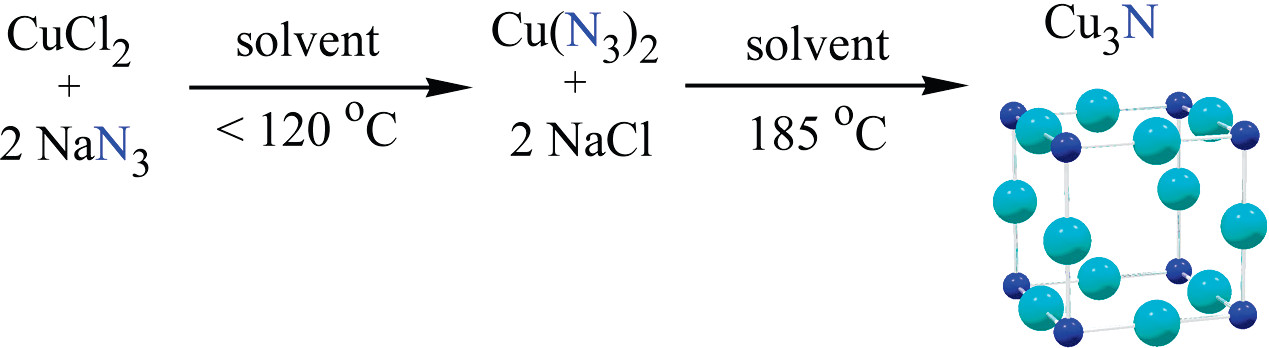





Leave a comment